
 扫码支付更轻松
扫码支付更轻松

导航栏各模块说明:
首页
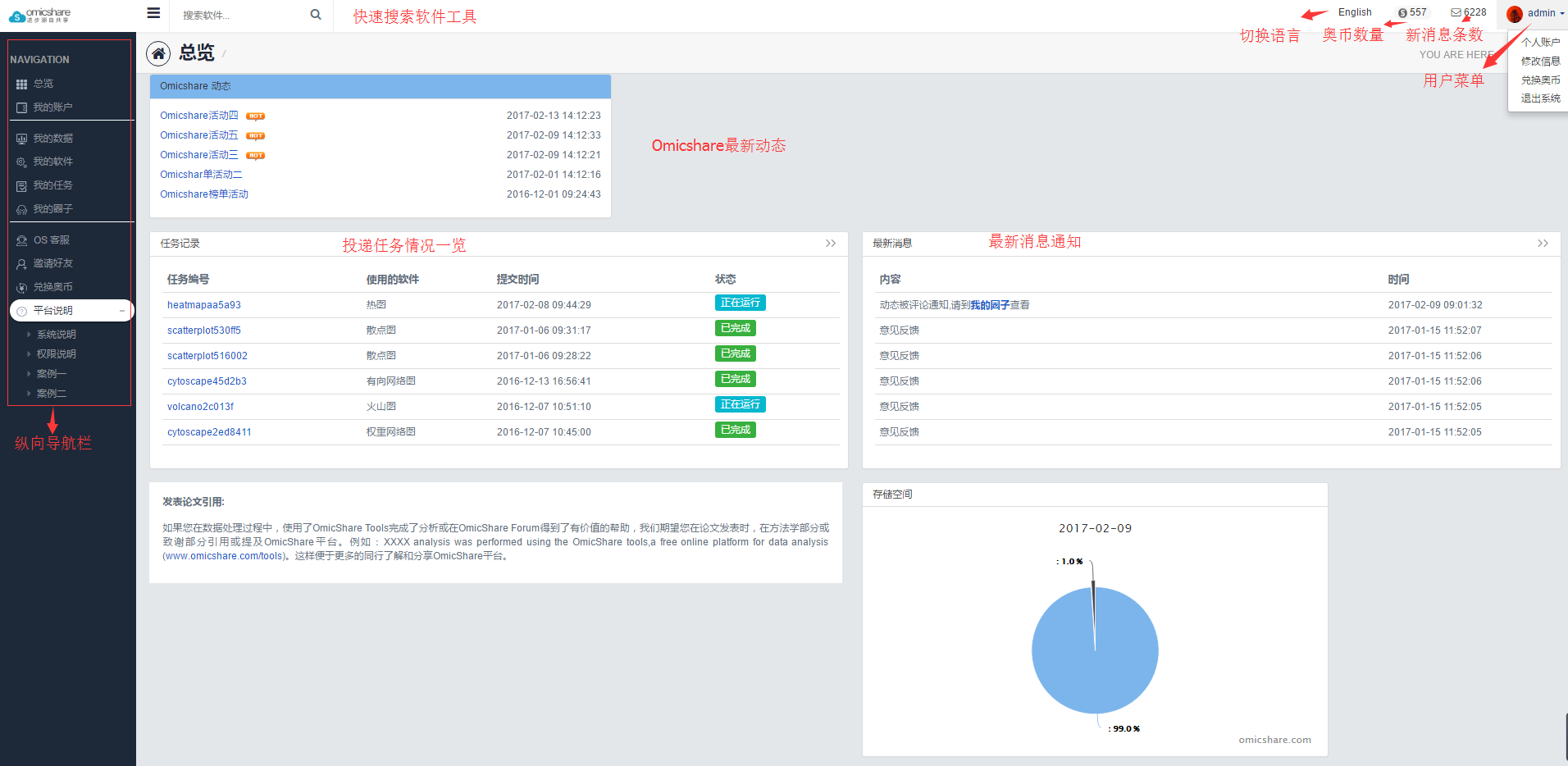
- 首页包括5个部分,一个是OmicShare动态,主要是近期OmicShare发布的动态。2、投递的项目状态,最新执行的项目的信息一览。3、我的消息,包含系统消息和个人消息,系统消息包含工具运行状态通知和系统推送消息。4、云平台引用说明。5、存储空间使用情况,用饼图显示存储空间的使用情况 。
工具库
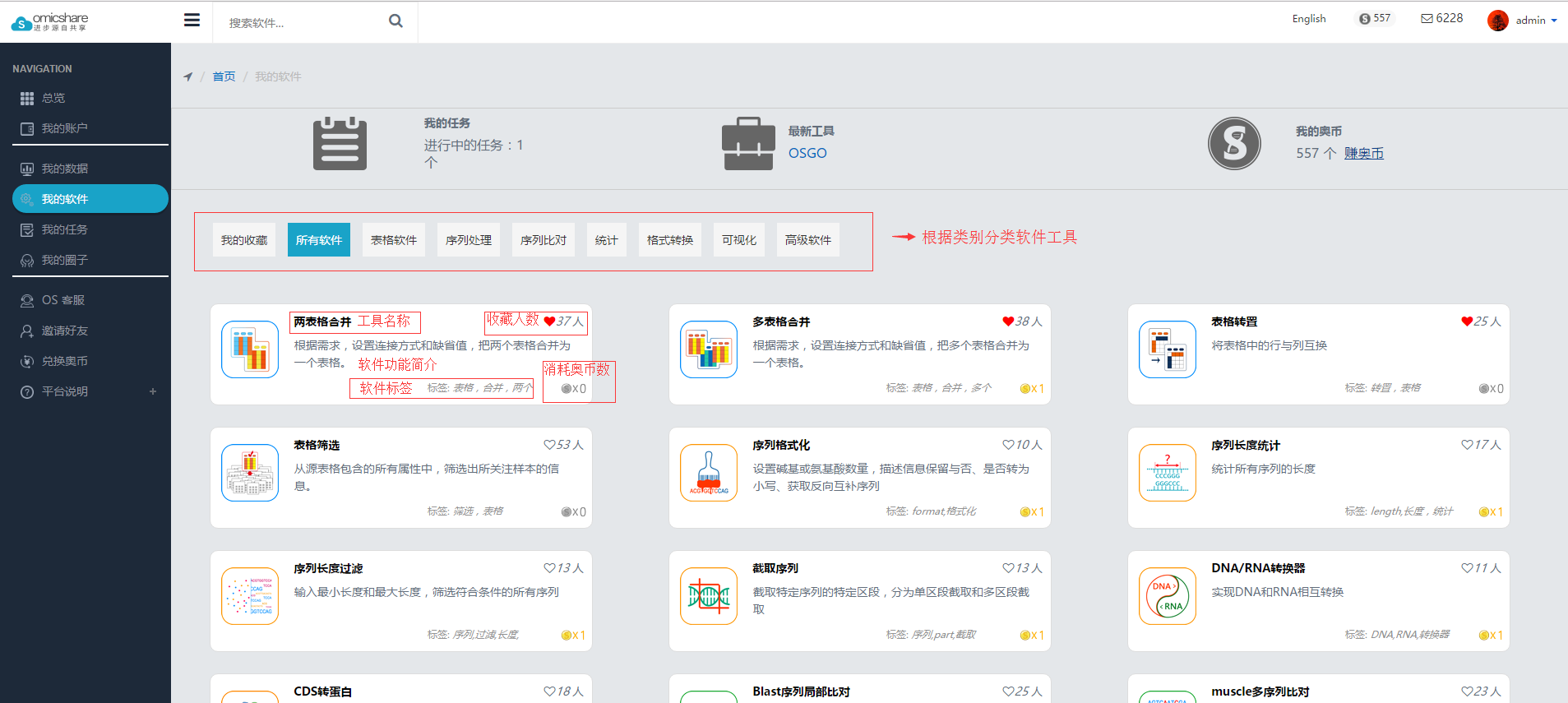
- 工具库包含五类基本生物工具工具:表格工具类,序列处理工具类、序列比对类、统计类、可视化图形类和格式转换类。另外还有高级工具,如趋势分析,pathway富集分析、go富集分析、OSGO分析等。
- 云平台目前共有57个生物工具工具可以使用,可以根据类别快速找到需要的工具。
- 用户可以收藏自己常用的工具,以方便快速查找使用。
我的文件
- 我的文件里储存了用户上传的所有文件,并有详细描述文件的上传时间和大小。用户可以选择按文件名、时间先后、文件大小或者是文件类型排列文件.
- 用户可以进行新建文件夹、文件上传与下载、移动文件位置、重命名文件、删除文件,也可以批量操作文件。另外,还可以按不同条件查找文件。注意:新建文件夹和重命名文件时不能出现特殊符号,否则操作无效。
- 另外建议,把要使用的文件先上传到云端,这样在使用工具时可以选择已上传的云端文件,省去了提交项目时本地文件反复上传效率低而且速度缓慢的麻烦。
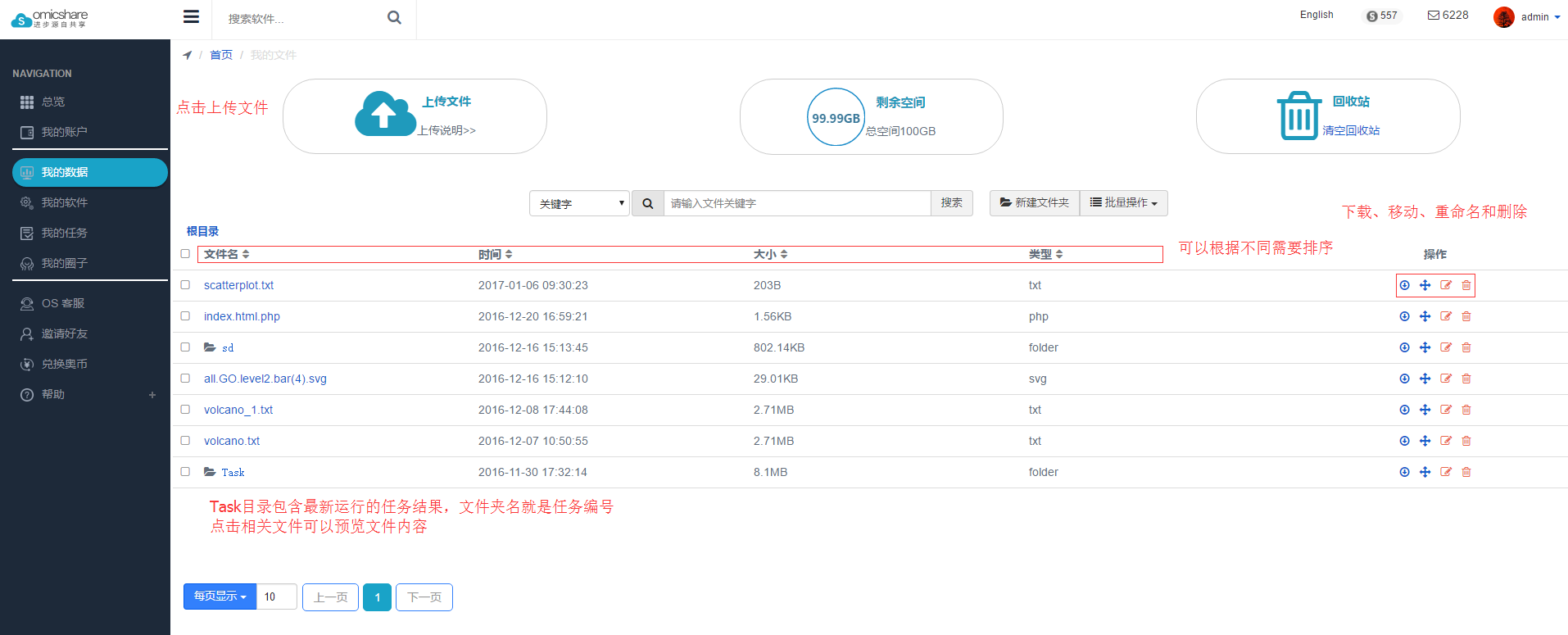
如图所示,页面中间有搜索框,可以对文件进行快速查找,可选择关键字查找、文件大小查找、文件类型查找、最后修改时间等不同方式进行查找。用户可以上传新文件,可以新建文件夹来管理文件,也可以批量下载、移动、删除文件。
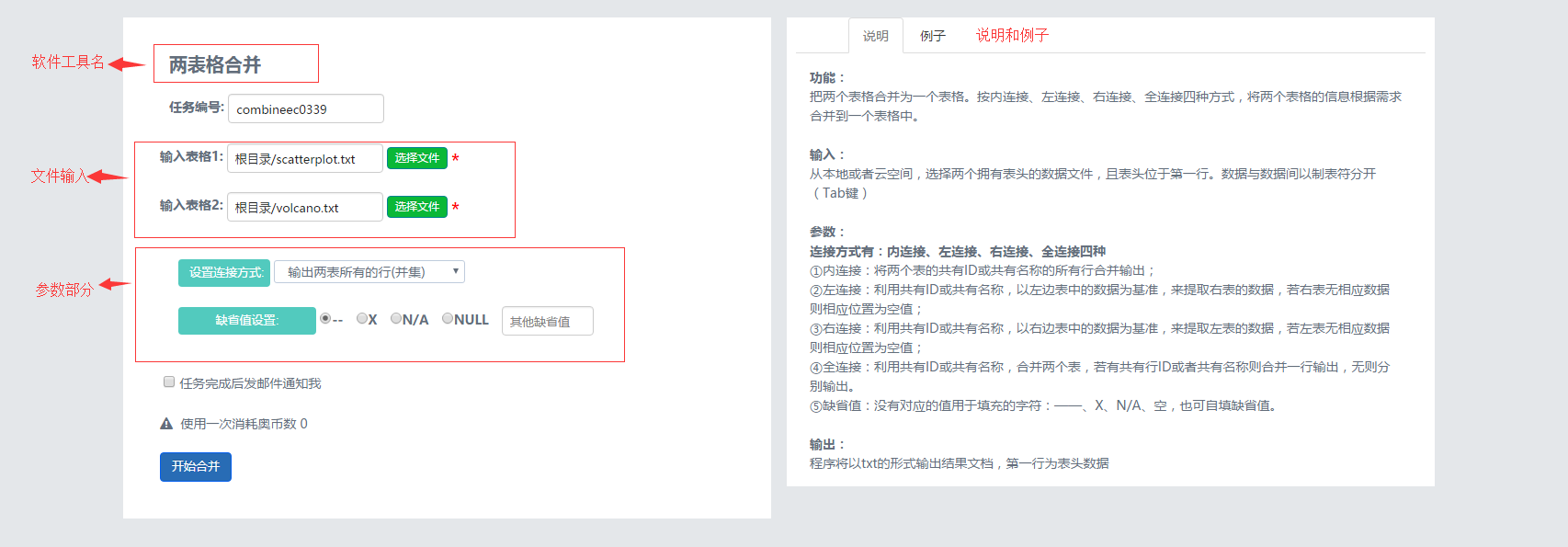
单个工具界面
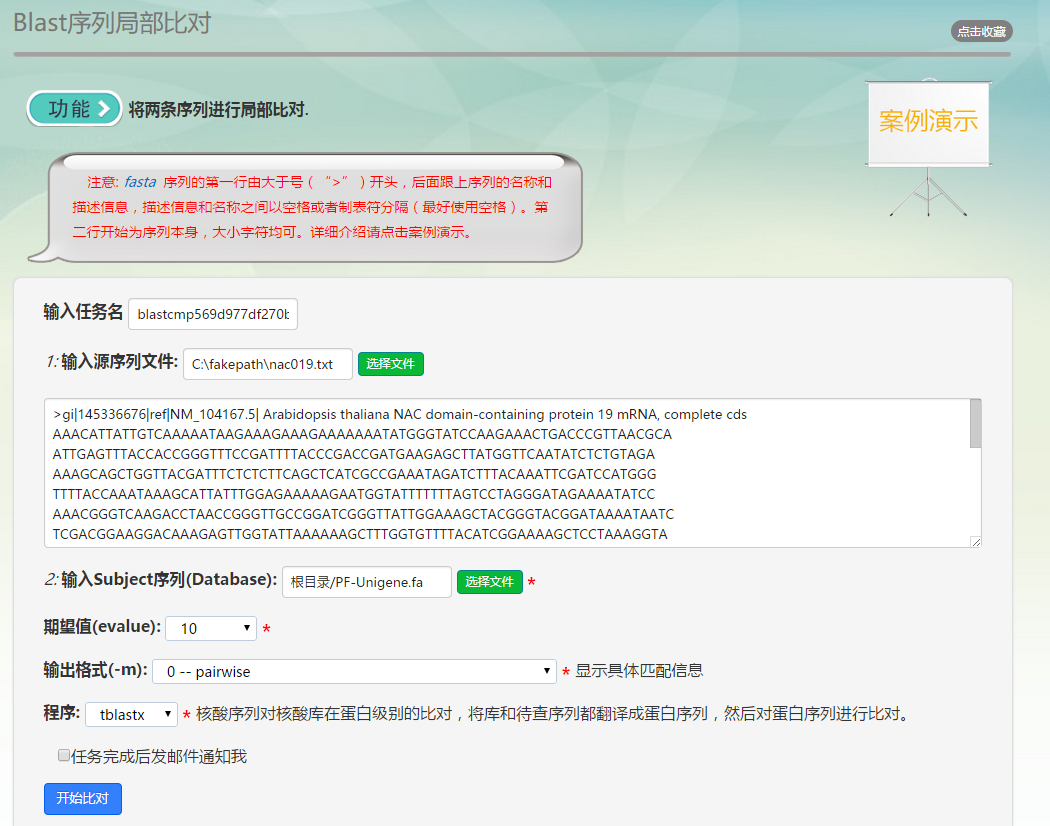
用户使用工具的界面。这里以两个表格合并 为例。
使用工具提交后,就会跳转到项目队列页面,用户提交生成的项目会在此排队。
我的项目
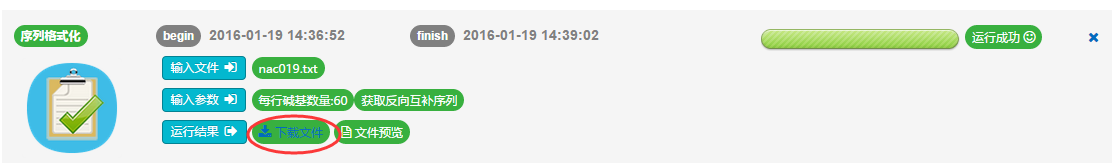
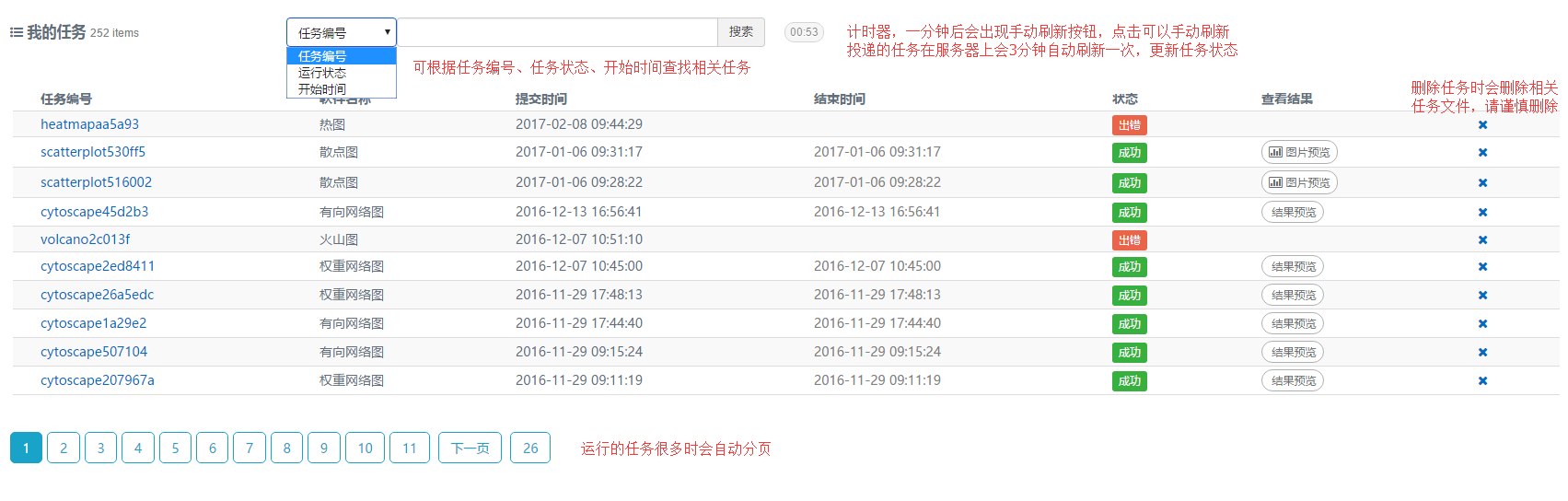
- 所有工具使用后都会生成对应的项目,项目状态包括失败,排队,运行,完成。用户可以通过关键字搜索相关项目,系统每隔3分钟自动刷新项目列表,用户也可以一分钟后手动刷新项目列表。项目的状态变为成功时,可以查看文件(缩略图)和下载结果文件。
- 在我的文件里也有Task文件夹,它会保存提交项目的结果文件,只需要按当时提交的项目名查找结果文件夹即可找到结果文件。
回收站
回收站里面显示的文件是已删除的文件,可还原也可彻底删除。还原文件就能恢复文件到用户个人根目录下,彻底删除则完全清除文件,不能再查找到文件的痕迹。回收站也支持批量删除与还原。
积分/奥币模块
①奥币数和论坛的奥币数同步,每次使用工具提交项目就会扣取一定奥币数,消耗的值在每个工具界面都有显示,奥币数太少,可能会不足以使用工具提交项目,此时可以登录论坛,在论坛有很多方式可以获取奥币。
附:使用工具奥币消耗规则
②关于积分,每天登录云平台即可获得1积分,连续登录系统一周额外赠送3积分,积分值与等级直接相关,用户等级分为三个等级,萌新、大师和VIP会员。鼠标放在等级图标上即可显示当前积分值。
新消息
- 新消息包含用户收到的所有信息,后面的数字是所有消息的总条数。点击可以查看所有系统消息和个人消息。
- 系统消息是自己的项目完成后的通知消息,或者是奖励消息,即额外增加积分/奥币的信息。
- 个人消息是管理员发送或回复的消息。一般是用户在网站发送反馈信息后,管理员的回复。
用户菜单
- 修改个人基本信息,如更新密码,电话、邮件。
- 兑换积分,用户获得积分码后,可以兑换获得积分,提升等级,获得更全面的功能体验。
- 退出网站。
在线客服
我的客服服务有四种方式,分别为加群讨论,论坛发帖、发送邮件和填写工单。推荐加群和论坛发帖,能最迅速的得到答案。




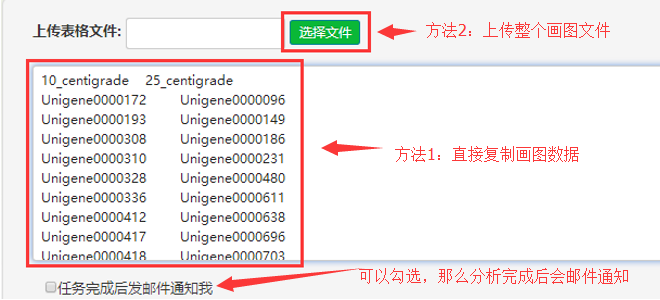 2).查看和下载分析结果
2).查看和下载分析结果

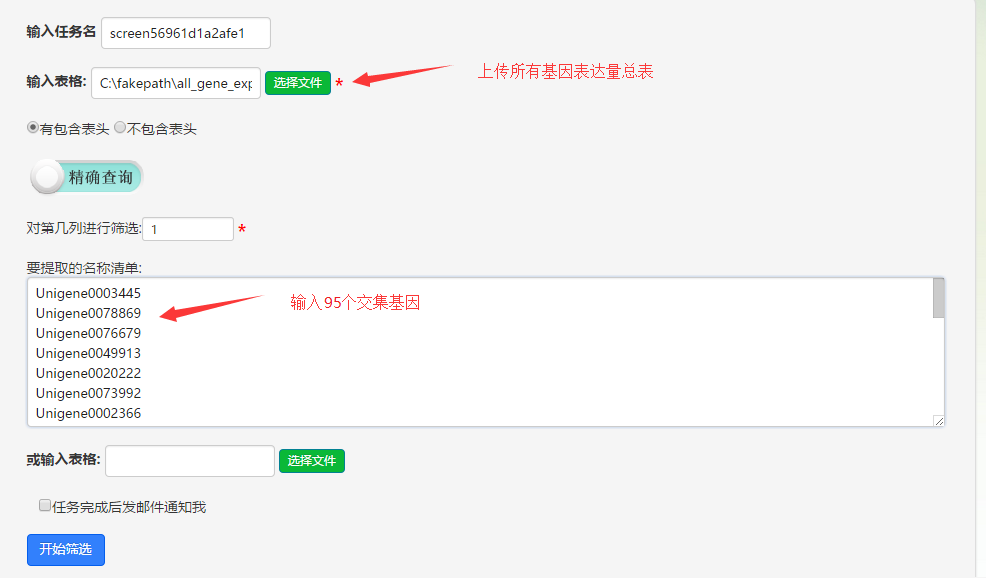
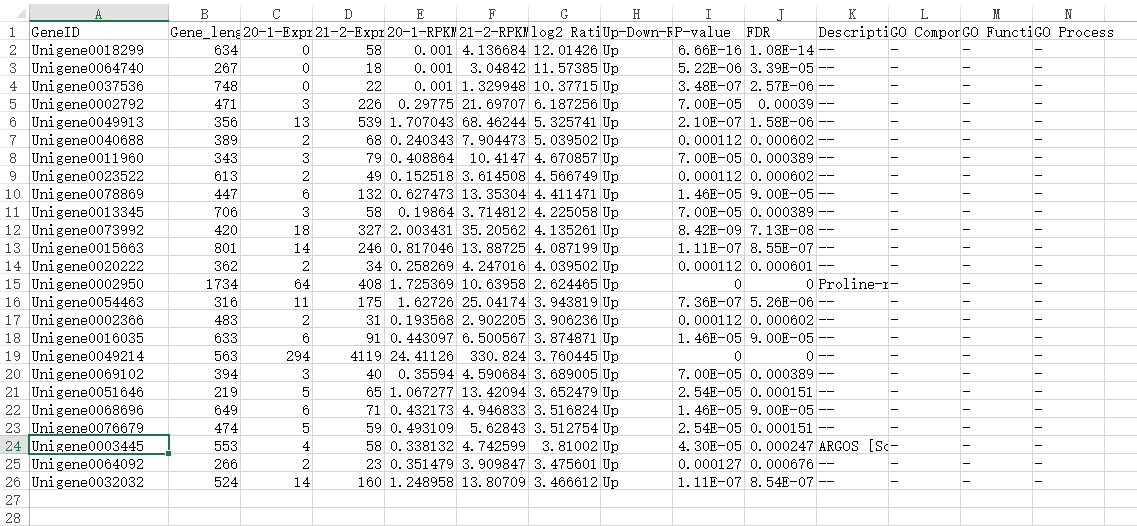
 2).查看并下载数据结果
2).查看并下载数据结果