



 扫码支付更轻松
扫码支付更轻松

功能:
这个实用程序使得根据其SAM标志值来识别读取的属性很容易,或者相反,为给定的属性组合找到SAM Flag值。
SAM文件格式:
sam文件格式包括两部分,头文件(header)和比对文件(alignment)。头文件描述sam文件是否排序及参考序列的信息,一般被隐藏起来;而比对文件一般描述比对的信息,且前11列是固定的,第12列是可变的,每列解释如下:
1 | QNAME | Query template NAME | reads名称
2 | FLAG | bitwise FLAG | 位标签
3 | RNAME | Reference sequence NAME | 参考序列名称
4 | POS | 1‐based leftmost POSition | 基于从1开始的左起比对位置
5 | MAPQ | MAPping Quality | reads比对质量
6 | CIAGR | CIGAR string | CIGAR字符串
7 | RNEXT | Ref. name of the mate/next read | 配对的reads比对到的参考序列名称
8 | PNEXT | Position of the mate/next read | 配对的reads比对到的位置
9 | TLEN | observed Template LENgth | reads与配对reads组成的片段的长度
10 | SEQ | segment SEQuence | reads序列
11 | QUAL | ASCII of Phred-scaled base QUALity+33 | 基于ASCII码-33质量体系的reads质量值
*12 | optional fields | variable OPTional fields in the format TAG:TYPE:VALUE
| 可选字段,格式为TAG:TYPE:VALUE,不同比对工具给出的字段及含义是不一样的
输入:
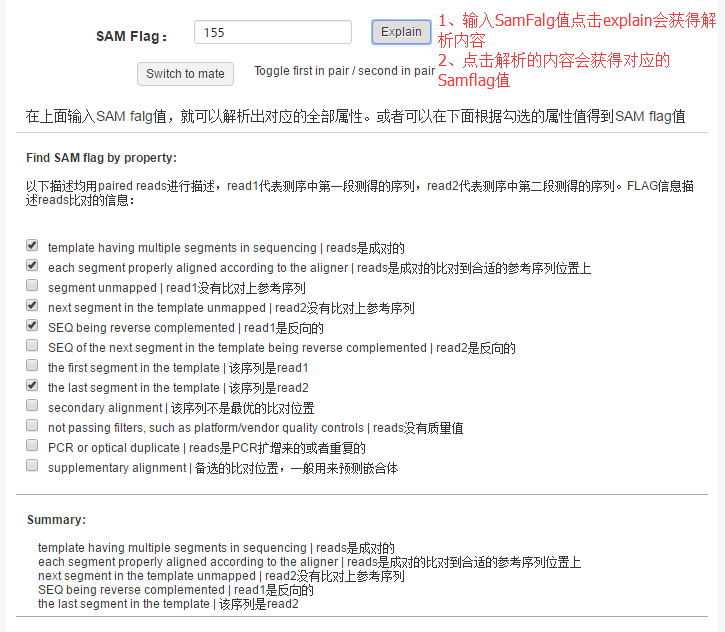
文件格式说明:在SAM flag框里输入要解释的正整数值.
参数:
① 0x1 template having multiple segments in sequencing | reads是成对的
② 0x2 each segment properly aligned according to the aligner | reads是成对的比对到合适的参考序列位置上
③ 0x4 segment unmapped | 该read没有比对上参考序列
④ 0x8 next segment in the template unmapped | 成对的另一端read没有比对上参考序列
⑤ 0x10 SEQ being reverse complemented | 该read是反向的
⑤ 0x20 SEQ of the next segment in the template being reverse complemented | 成对的另一端read是反向的
⑥ 0x40 the first segment in the template | 该序列是read1
⑦ 0x80 the last segment in the template | 该序列是read2
⑧ 0x100 secondary alignment | 该序列不是最优的比对位置
⑨ 0x200 not passing filters, such as platform/vendor quality controls | reads没有质量值
⑩ 0x400 PCR or optical duplicate | reads是PCR扩增来的或者重复的
⑪ 0x800 supplementary alignment | 备选的比对位置,一般用来预测嵌合体
输出:
工具结果将输出SAM flag的解析结果。
Q1. 上传的数据需要保存成什么格式?文件名称和拓展名有没有要求?
OmicShare当前支持txt(制表符分隔)文本文件、csv(逗号分隔)文本文件、以及Excel专用的xlsx格式,同样支持旧版Excel的xls(Excel 97-2003 )格式。如果是核酸、蛋白序列文件,必须为FASTA格式(本质是文本文件)。
文件名可由英文和数字构成,文件拓展名没有限制,可以是“.txt”、“.xlsx”、“.xls”、“.csv”“.fasta”等,例如 mydata01.txt,gene02.xlsx 。
Q2. 提交时报错常见问题:
1.提交时显示X行X列空行/无数据,请先自查表格中是否存在空格或空行,需要删掉。
2.提交时显示列数只有1列,但表格数据不止1列:列间需要用分隔符隔开,先行检查文件是否用了分隔符。
其它提示报错,请先自行根据提示修改;如果仍然无法提交,可通过左侧导航栏的“联系客服”选项咨询OmicShare客服。
Q3. 提交的任务完成后却不出图该怎么办?
主要原因是上传的数据文件存在特殊符号所致。可参考以下建议逐一排查出错原因:
(1)数据中含中文字符,把中文改成英文;
(2) 数据中含特殊符号,例如 %、NA、+、-、()、空格、科学计数、罗马字母等,去掉特殊符号,将空值用数字“0”替换;
(3)检查数据中是否有空列、空行、重复的行、重复的列,特别是行名(一般为gene id)、列名(一般为样本名)出现重复值,如果有删掉。
排查完之后,重新上传数据、提交任务。如果仍然不出图,可通过左侧导航栏的“联系客服”选项咨询OmicShare客服。
Q4.下载的结果文件用什么软件打开?
OmicShare云平台的结果文件(例如,下图为KEGG富集分析的结果文件)包括两种类型:图片文件和文本文件。

图片文件:
为了便于用户对图片进行后期编辑,OmicShare同时提供位图(png)和矢量图(pdf、svg)两种类型的图片。对于矢量图,最常见的是pdf和svg格式,常用Ai(Adobe illustrator)等进行编辑。其中,svg格式的图片可用网页浏览器打开,也可直接在word、ppt中使用。
文本文件:
文本文件的拓展名主要有4种类型:“.os”、“.xls”、“.log”和“.txt”。这些文件本质上都是制表符分隔的文本文件,使用记事本、Notepad++、EditPlus、Excel等文本编辑器直接打开即可。结果文件中,拓展名为“.os”文件为上传的原始数据;“.xls”文件一般为分析生成的数据表格;“.log”文件为任务运行日志文件,便于检查任务出错原因。
Q5. 提交的任务一直在排队怎么办?
提交任务后都需要排队,1分钟后,点击“任务状态刷新”按钮即可。除了可能需运行数天的注释工具,一般工具数十秒即可出结果,如果超出30分钟仍无结果,请联系OS客服,发送任务编号给OmicShare客服,会有专人为你处理任务问题。
Q6. 结果页面窗口有问题,图表加载不出来怎么办?
尝试用谷歌浏览器登录OmicShare查看结果文件,部分浏览器可能不兼容。