


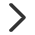

 扫码支付更轻松
扫码支付更轻松

1. 功能:O2PLS是一种泛化的OPLS,可在两个数据矩阵中进行双向建模和预测,利用此分析,可挖掘两组学之间的内部联系,确定两组学数据的关联程度,同时确定引起这种关联的主要基因或者代谢物或蛋白等。
2. 应用:蛋白+代谢 / 蛋白+脂质 / 转录+代谢 / 转录+脂质 / 16S+代谢等两组学关联分析。
3. 输入:两个数据矩阵,格式txt,xlsx,csv
文件1:如转录组表达量数据(注意:输入数据不能少于6个样本)
| gene id | CK1 | CK2 | CK3 | Work1 | Work2 | Work3 |
| unigene002693 | 0 | 0 | 2.53 | 1.96 | 0.34 | 0.096 |
| unigene003854 | 0.52 | 0.12 | 0.58 | 0.12 | 0 | 0.22 |
| …… | …… | …… | …… | …… | …… | …… |
文件2:如代谢组丰度数据(注意:输入数据不能少于6个样本)
| metabolite | CK1 | CK2 | CK3 | Work1 | Work2 | Work3 |
| var_1 | 20.623 | 128.61 | 154.6 | 41.458 | 227.88 | 113.78 |
| var_2 | 15.682 | 67.41 | 36.14 | 67.913 | 71.591 | 45.11 |
| …… | …… | …… | …… | …… | …… | …… |
4. 参数
颜色:根据需求选择荷载图中散点颜色
字体大小:根据需求选择荷载图中字体大小
散点大小:根据需求选择荷载图中散点大小
散点透明度:根据需求选择荷载图中散点的透明度
显示标签:根据需求选择荷载图中要展示的标签数量
5. 输出:
(1) 模型构建参数:
| ax | ay | a | prediction error |
ax:转录组正交部分的组分个数
ay:代谢组正交部分的组分个数
a:两组学关联部分的组分个数
prediction error:模型的预测误差
交叉验证法通过多次预建模,选择预测误差(prediction error)最小的模型进行后续分析。
(2) 贡献度评估
| R2X | R2Y | R2Xcor | R2Ycor |
R2x:转录组关联和正交部分对转录组总变异的解释度
R2y:代谢物关联和正交部分对代谢组总变异的解释度
R2xcorr:转录组关联部分对转录组总变异的解释度
R2ycorr:代谢物关联部分对代谢组总变异的解释度
贡献度是指模型各部分对总变异的解释程度,用R2表示。R2值越高,表示该部分对模型的解释能力越好。
(3) loading分值表
载荷值(loading value)表示变量(代谢物/基因)在各组分的解释能力(即对组间差异的贡献度),载荷值的正负表示与另一组学正关联或负相关;载荷值的绝对值越大,表示关联越强。
| metabolite | Loading_1 | Loading_2 |
| POS0052 | ||
| POS02378 |
| transcript | Loading_1 | Loading_2 |
| gene1 | ||
| gene2 |
Loading_1:一维loading值
Loading_2:二维loading值
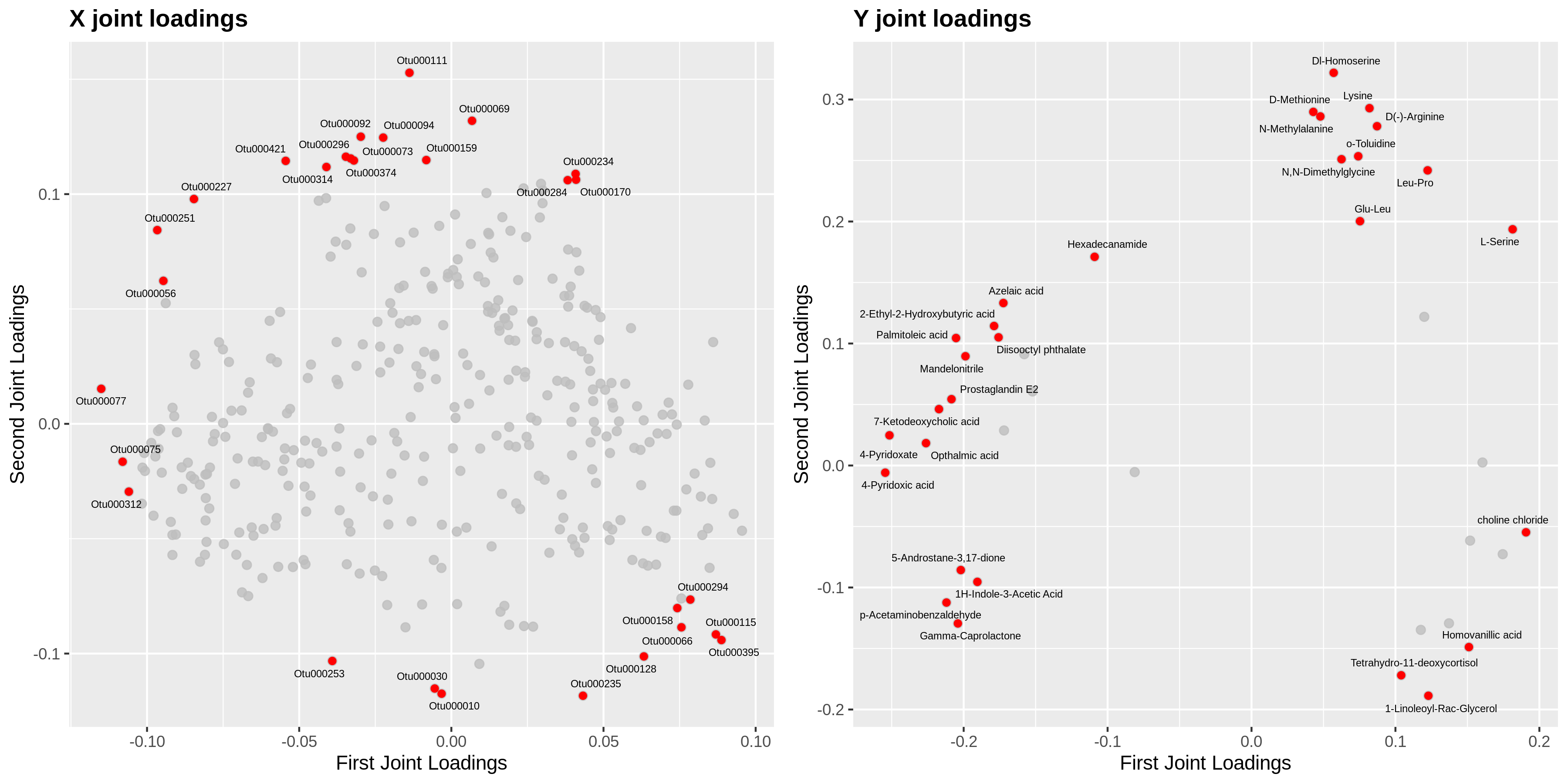
(4) 两组学o2pls载荷图
为了查看哪些代谢物和基因相互关联,我们对关联的部分变量(基因或代谢物)分别绘制不同组学的载荷图(loadings plot)
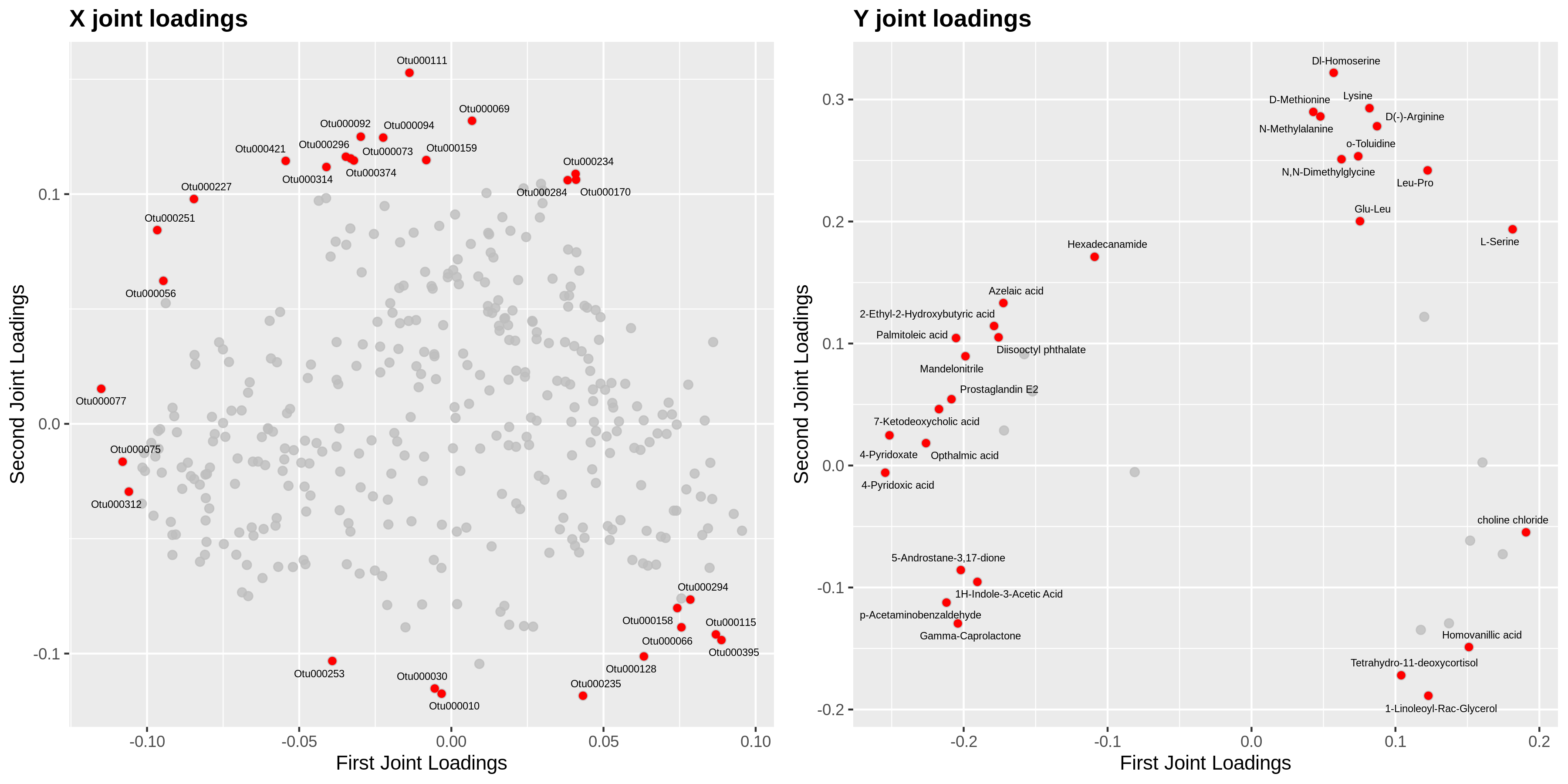
图1 两组学O2PLS荷载图
(5) 两组学关联载荷图
根据元素loading值结果,我们筛选出前两个维度loading值平方和前n个基因和代谢物进行整合绘制loading图,以展示关联程度最大的基因和代谢物。(这里的n在显示标签提前设置)
图2 两组学关联荷载图
该图表示转录组和代谢物关联部分的载荷图,横轴为第一维坐标,纵轴为第二维坐标。图中圆点表示基因或代谢物,在坐标中的绝对值越大,表示此元素与另外一个组学的关联程度越大。从该图中,我们就可以获得关联性较高的基因和代谢物,为后续的两组学数据关联分析指明方向。
使用16S数据与代谢组关联分析为例:
示例数据
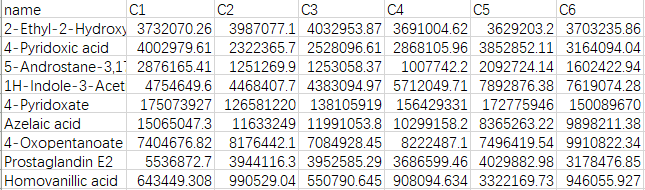
代谢组数据(展示部分)
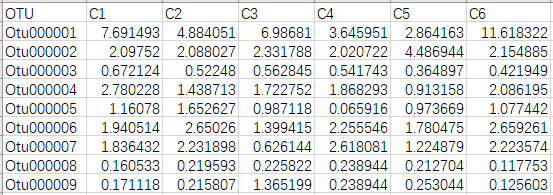
16S数据(展示部分)
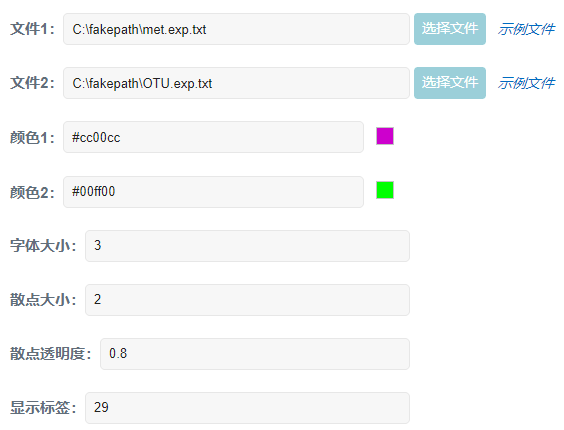
参数设置:
输出结果:
(1)模型构建参数文件,以cv.stat.xls命名
交叉验证法通过多次预建模,选择预测误差(prediction error)最小的模型进行后续分析。
![]()
(2)贡献度评估,以component_proportions.xls命名
贡献度是指模型各部分对总变异的解释程度,用R2表示。R2值越高,表示该部分对模型的解释能力越好。本例子中解释度为58.8%和65.4%,说明模型较好。
![]()
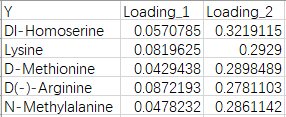
(3)loading分值表,以X_loading.xls和Y_loading.xls命名
载荷值(loading value)表示变量(代谢物/基因)在各组分的解释能力(即对组间差异的贡献度),载荷值的正负表示与另一组学正关联或负相关;载荷值的绝对值越大,表示关联越强。
(4)两组学o2pls载荷图,以o2m_loading.pdf和o2m_loading.png命名,提供pdf和png格式
查看微生物种和代谢物关联的数据,用o2pls荷载图展示。
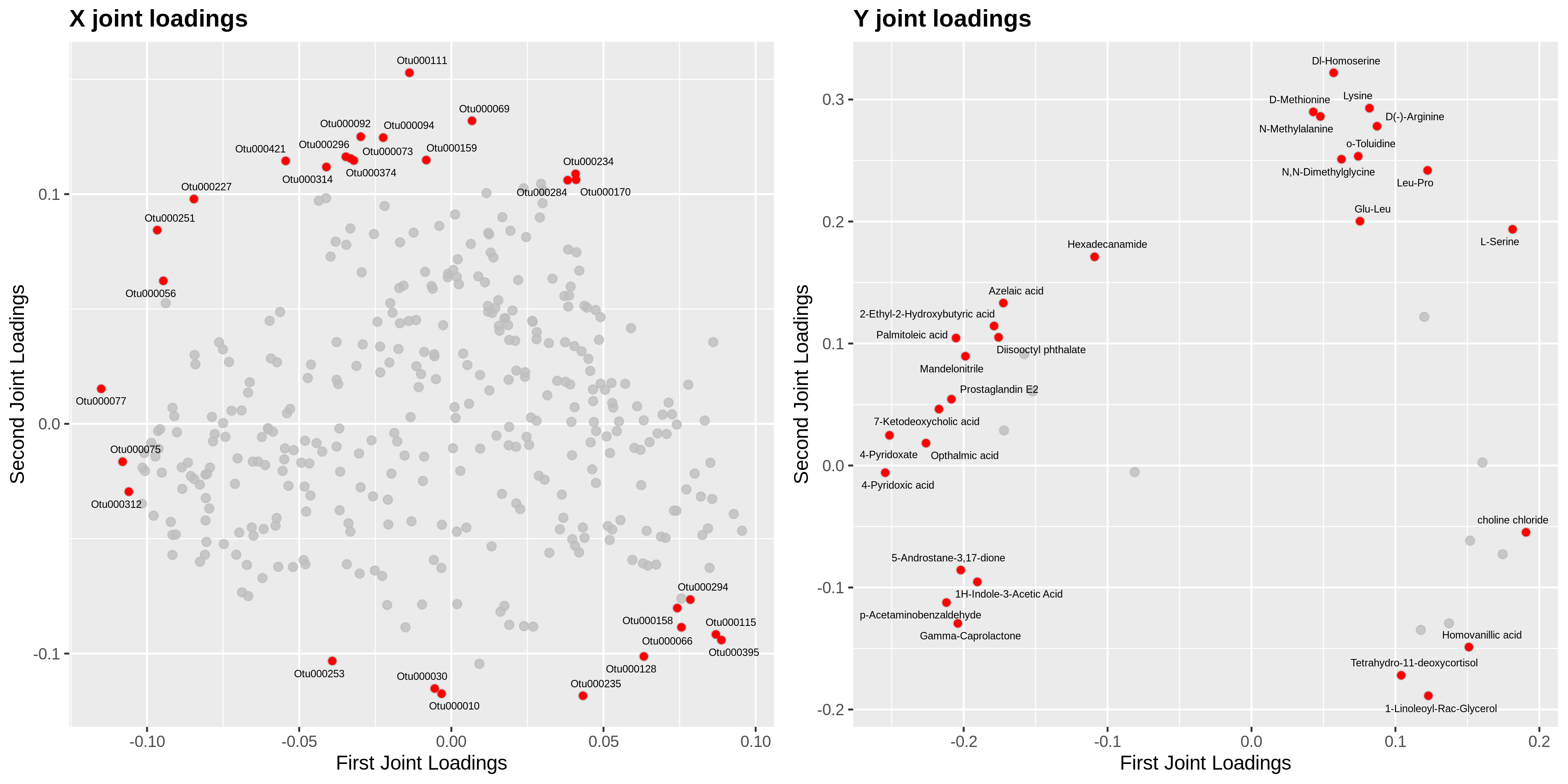
图1 两组学O2PLS荷载图
5. 两组学关联载荷图
从该图中,我们就可以获得关联性较高的微生物OTU和分泌代谢物,为后续的两组学数据关联分析指明方向。
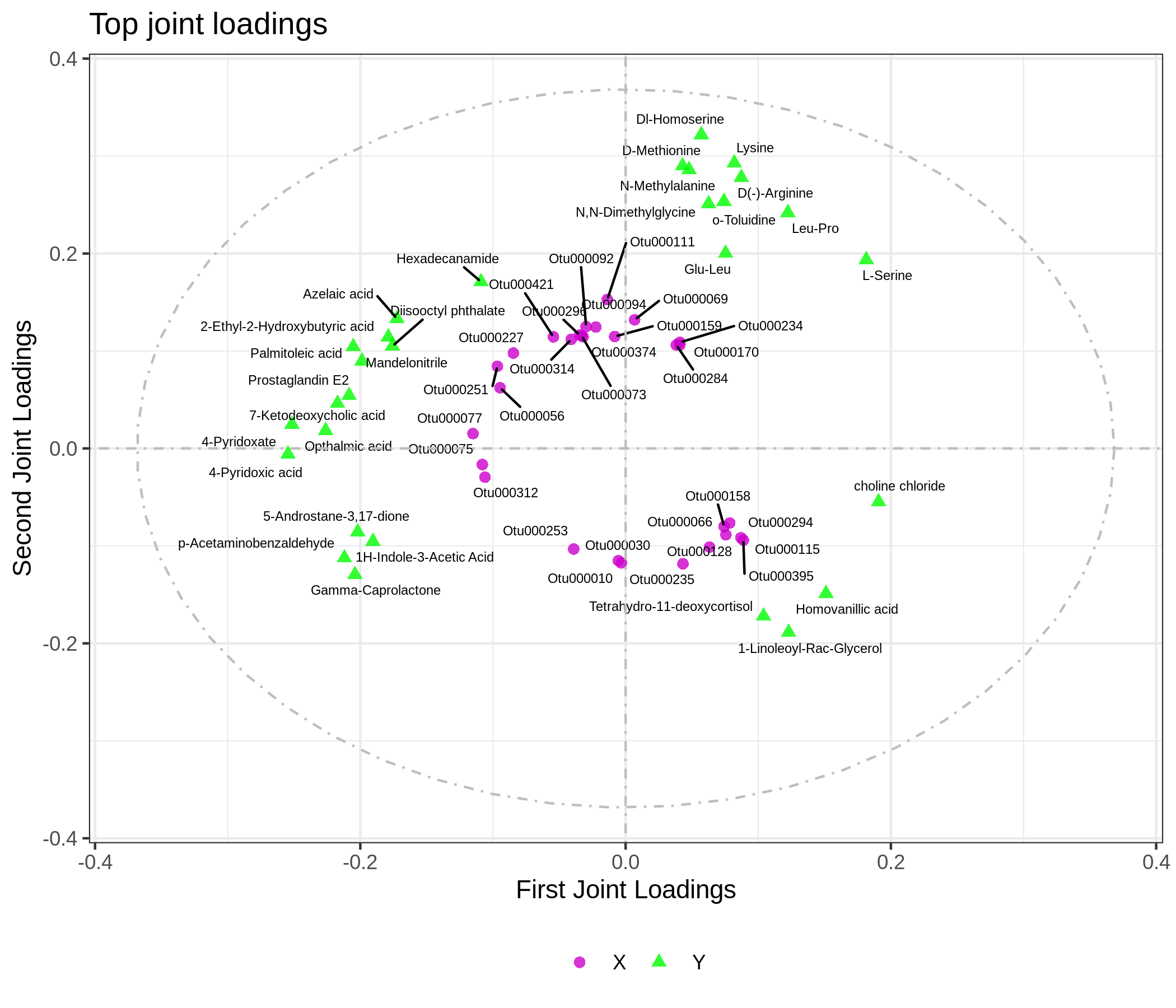
图2 两组学关联荷载图
Q1. 上传的数据需要保存成什么格式?文件名称和拓展名有没有要求?
OmicShare当前支持txt(制表符分隔)文本文件、csv(逗号分隔)文本文件、以及Excel专用的xlsx格式,同样支持旧版Excel的xls(Excel 97-2003 )格式。如果是核酸、蛋白序列文件,必须为FASTA格式(本质是文本文件)。
文件名可由英文和数字构成,文件拓展名没有限制,可以是“.txt”、“.xlsx”、“.xls”、“.csv”“.fasta”等,例如 mydata01.txt,gene02.xlsx 。
Q2. 提交时报错常见问题:
1.提交时显示X行X列空行/无数据,请先自查表格中是否存在空格或空行,需要删掉。
2.提交时显示列数只有1列,但表格数据不止1列:列间需要用分隔符隔开,先行检查文件是否用了分隔符。
其它提示报错,请先自行根据提示修改;如果仍然无法提交,可通过左侧导航栏的“联系客服”选项咨询OmicShare客服。
Q3. 提交的任务完成后却不出图该怎么办?
主要原因是上传的数据文件存在特殊符号所致。可参考以下建议逐一排查出错原因:
(1)数据中含中文字符,把中文改成英文;
(2) 数据中含特殊符号,例如 %、NA、+、-、()、空格、科学计数、罗马字母等,去掉特殊符号,将空值用数字“0”替换;
(3)检查数据中是否有空列、空行、重复的行、重复的列,特别是行名(一般为gene id)、列名(一般为样本名)出现重复值,如果有删掉。
排查完之后,重新上传数据、提交任务。如果仍然不出图,可通过左侧导航栏的“联系客服”选项咨询OmicShare客服。
Q4.下载的结果文件用什么软件打开?
OmicShare云平台的结果文件(例如,下图为KEGG富集分析的结果文件)包括两种类型:图片文件和文本文件。

图片文件:
为了便于用户对图片进行后期编辑,OmicShare同时提供位图(png)和矢量图(pdf、svg)两种类型的图片。对于矢量图,最常见的是pdf和svg格式,常用Ai(Adobe illustrator)等进行编辑。其中,svg格式的图片可用网页浏览器打开,也可直接在word、ppt中使用。
文本文件:
文本文件的拓展名主要有4种类型:“.os”、“.xls”、“.log”和“.txt”。这些文件本质上都是制表符分隔的文本文件,使用记事本、Notepad++、EditPlus、Excel等文本编辑器直接打开即可。结果文件中,拓展名为“.os”文件为上传的原始数据;“.xls”文件一般为分析生成的数据表格;“.log”文件为任务运行日志文件,便于检查任务出错原因。
Q5. 提交的任务一直在排队怎么办?
提交任务后都需要排队,1分钟后,点击“任务状态刷新”按钮即可。除了可能需运行数天的注释工具,一般工具数十秒即可出结果,如果超出30分钟仍无结果,请联系OS客服,发送任务编号给OmicShare客服,会有专人为你处理任务问题。
Q6. 结果页面窗口有问题,图表加载不出来怎么办?
尝试用谷歌浏览器登录OmicShare查看结果文件,部分浏览器可能不兼容。