



 扫码支付更轻松
扫码支付更轻松

功能:输入蛋白序列,根据序列或结构差异关系绘制进化树,序列比对使用 Muscle 工具,最大似然法构树使用 iqtree 工具,比邻法构树使用 fastme 工具。
序列文件:只允许输入蛋白序列(fasta格式,最多100条蛋白序列,单个蛋白序列长度不超过5000个氨基酸)
>AtSWEET1
MNIAHTIFGVFGNATALFLFLAPSITFKRIIKNKSTEQFSGIPYPMTLLNCLLSAWYGLPFVSKDNTLVSTINGTGAVIETVYVLI
FLFYAPKKEKIKIFGIFSCVLAVFATVALVSLFALQGNGRKLFCGLAATVFSIIMYASPLSIMRLVVKTKSVEFMPFFLSLFVFLCG
TSWFVYGLIGRDPFVAIPNGFGCALGTLQLILYFIYCGNKGEKSADAQKDEKSVEMKDDEKKQNVVNGKQDLQV
提示:假如输入36条蛋白序列,每条长度600个氨基酸,ML法构树大概要运行30-60分钟,而NJ法大概需要3-10分钟,大家把握时间,耐心等待;如果运行时间超过1.5个小时,可联系客服。
参数:
建树方法: 建树需要根据自己的研究内容选择合适的建树方法。一般来说,若有合适的分子进化模型可供选择,则用最大似然法构树获得的结果较好;对于远缘物种序列,一般使用邻接法或最大似然法。
Bootstrap: 即自展值,是用来检验你所计算的进化树分支可信度的。简单地讲就是把序列的位点都重排,重排后的序列再用相同的办法构树,如果原来树的分枝在重排后构的树中也出现了,就给这个分枝打上一分,如果没出现就给0分,这样经过你给定的repetitions次(至少1000次)重排构树打分后,每个分枝就都得出分值,计算机会给你换算成bootstrap值。重排的序列有很多组合,值越小说明分枝的可信度越低,最好根据数据的情况选用不同的构树方法和模型。
输出:
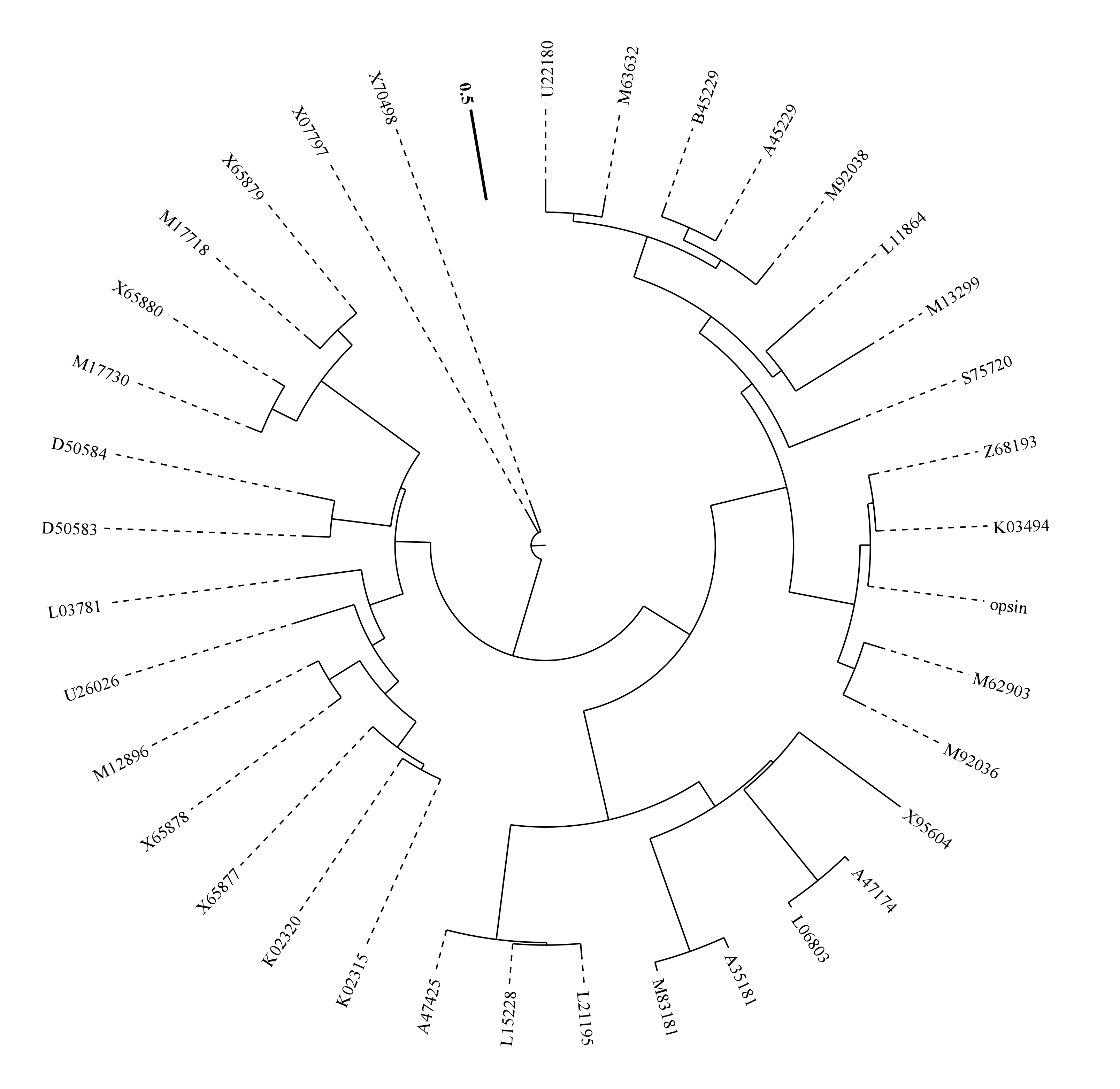
1. 分别生成png/svg格式的圈图和无根进化树
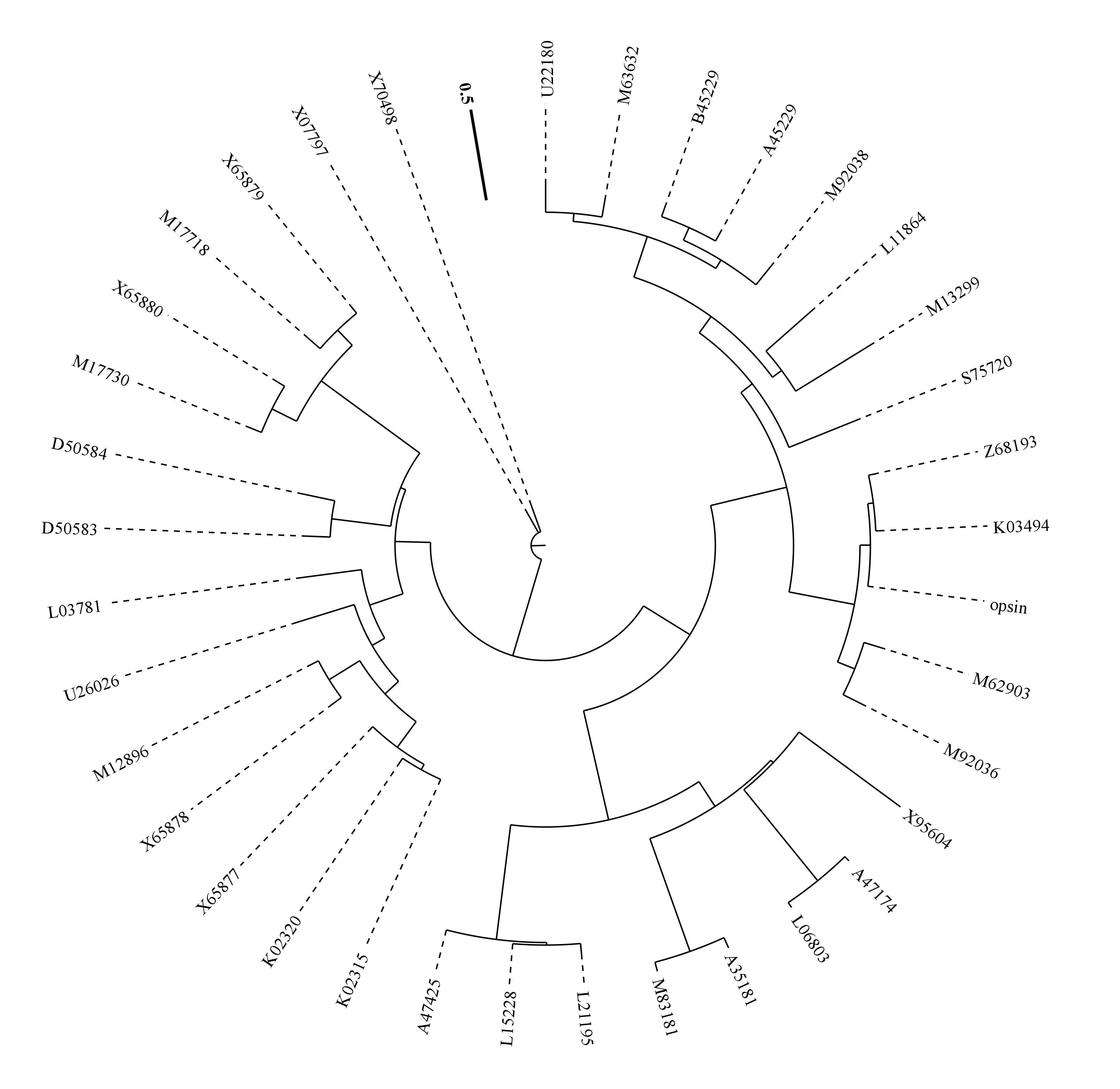
图1 圈图进化树
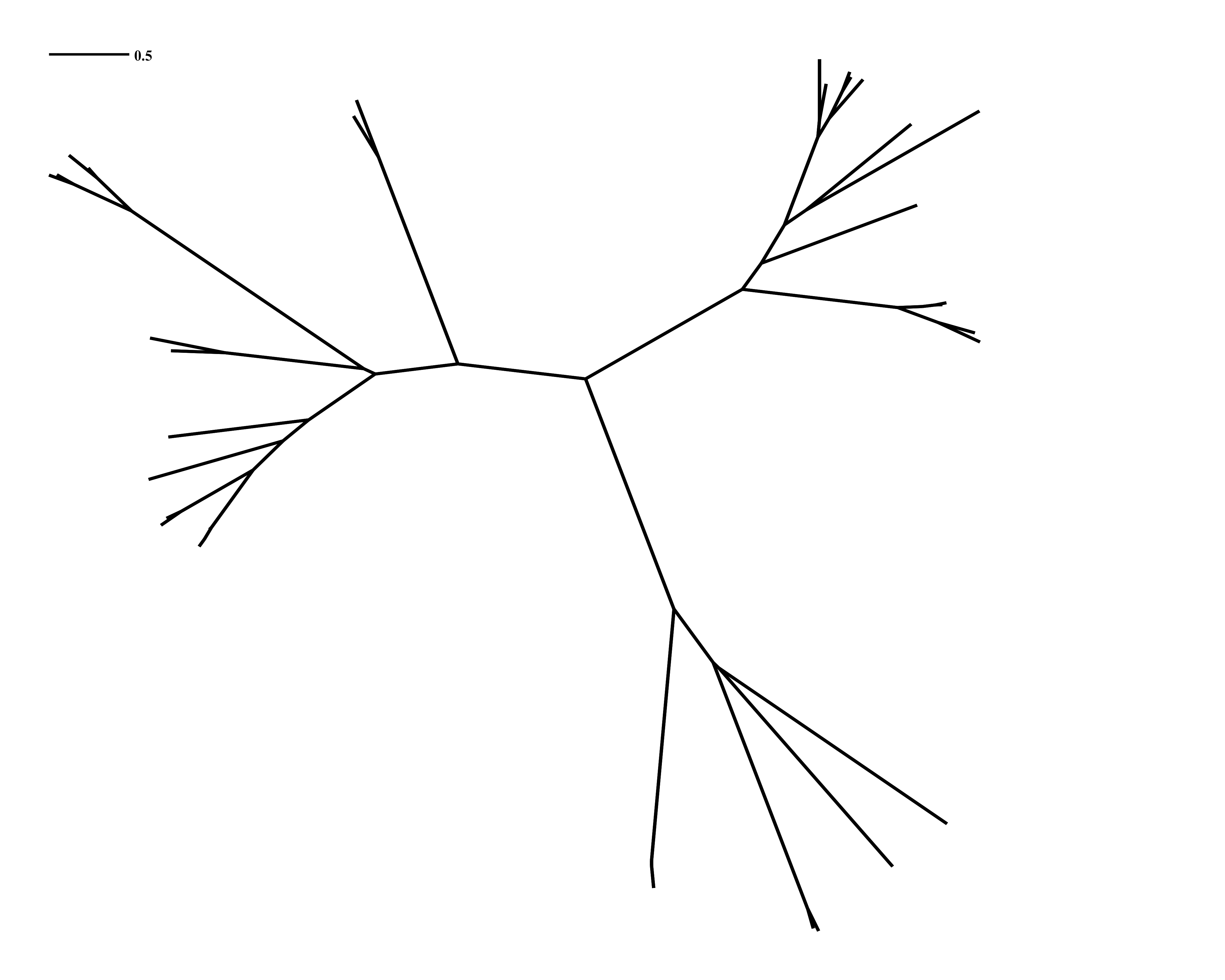
图2 无根进化树
2. nwk文件,方便用户用其他工具进行个性化修改,如利用iTOL (https://itol.embl.de/)对进化树美化。
![]()
3. 如果选择邻接法的输出文件如下:
result.msa.boot.txt 是bootstrap的每棵树的记录
result.msa.dist.xls 是每棵树的距离矩阵
result.msa.fa 是比对后的fasta格式(重要文件)
result.msa.nwk 是软件输出来的原始结果,不重要,可忽略
result.msa.phy 是比对后的fasta格式(重要文件)
result.nwk 是最终结果,方便用户用.nwk文件利用其他工具美化进化树
4. 如果选择最大似然法的输出文件如下:
result.msa.fa 是比对结果(重要文件)
result.msa.phy 是比对结果(重要文件)
result.msa.phy.bionj 是起始树,用BioNJ的方法构建的
result.msa.phy.ckp.gz 是构建ML树的软件的checkpoint文件,就是记录进行到哪一步了
result.msa.phy.contree 是bootstrap的一致树
result.msa.phy.iqtree 是用文本的形式展示了进化树的结构
result.msa.phy.log 是软件运行的日志文件
result.msa.phy.mldist 是ML距离矩阵
result.msa.phy.splits.nex 是bootstrap支持度的细化展示
result.msa.phy.treefile 是最终结果
result.nwk 是.treefile的拷贝文件,方便用户用.nwk文件利用其他工具美化进化树
5. 其他文件可以忽略,是程序运行文件。
注:对于进化树的美化,推荐使用iTOL (https://itol.embl.de/)美化,该工具功能齐全,也可以使用mega美化进化树,但功能有限。
例子:对拟南芥SWEET家族蛋白进行构树分析
1. 输入:
下面展示部分,详情点击示例文件链接
>AtSWEET1
MNIAHTIFGVFGNATALFLFLAPSITFKRIIKNKSTEQFSGIPYPMTLLNCLLSAWYGLPFVSKDNTLVSTINGTGAVIETVYVLIFLFYAPKKEKIKIF
GIFSCVLAVFATVALVSLFALQGNGRKLFCGLAATVFSIIMYASPLSIMRLVVKTKSVEFMPFFLSLFVFLCGTSWFVYGLIGRDPFVAIPNGFGCALGT
LQLILYFIYCGNKGEKSADAQKDEKSVEMKDDEKKQNVVNGKQDLQV
>AtSWEET6
MVHEQLNLIRKIVGILGNFISLCLFLSPTPTFIHIVKKKSVEKYSPLPYLATLLNCLVRALYGLPMVHPDSTLLVTISGIGITIEIVFLTIFFVFCGRQQ
HRLVISAVLTVQVVFVATLAVLVLTLEHTTDQRTISVGIVSCVFNAMMYASPLSVMKMVIKTKSLEFMPFLLSVVGFLNAGVWTIYGFVPFDPFLAIPNG
IGCVFGLVQLILYGTYYKSTKGIMEERKNRLGYVGEVGLSNAIAQTEPENIPYLNKRVSGV
>AtSWEET9
MFLKVHEIAFLFGLLGNIVSFGVFLSPVPTFYGIYKKKSSKGFQSIPYICALASATLLLYYGIMKTHAYLIISINTFGCFIEISYLFLYILYAPREAKIS
TLKLIVICNIGGLGLLILLVNLLVPKQHRVSTVGWVCAAYSLAVFASPLSVMRKVIKTKSVEYMPFLLSLSLTLNAVMWFFYGLLIKDKFIAMPNILGFL
FGVAQMILYMMYQGSTKTDLPTENQLANKTDVNEVPIVAVELPDVGSDNVEGSVRPMK
2. 参数设置:
建树方法:选择“最大似然法”
bootstrap:默认“1000”
3. 结果输出:
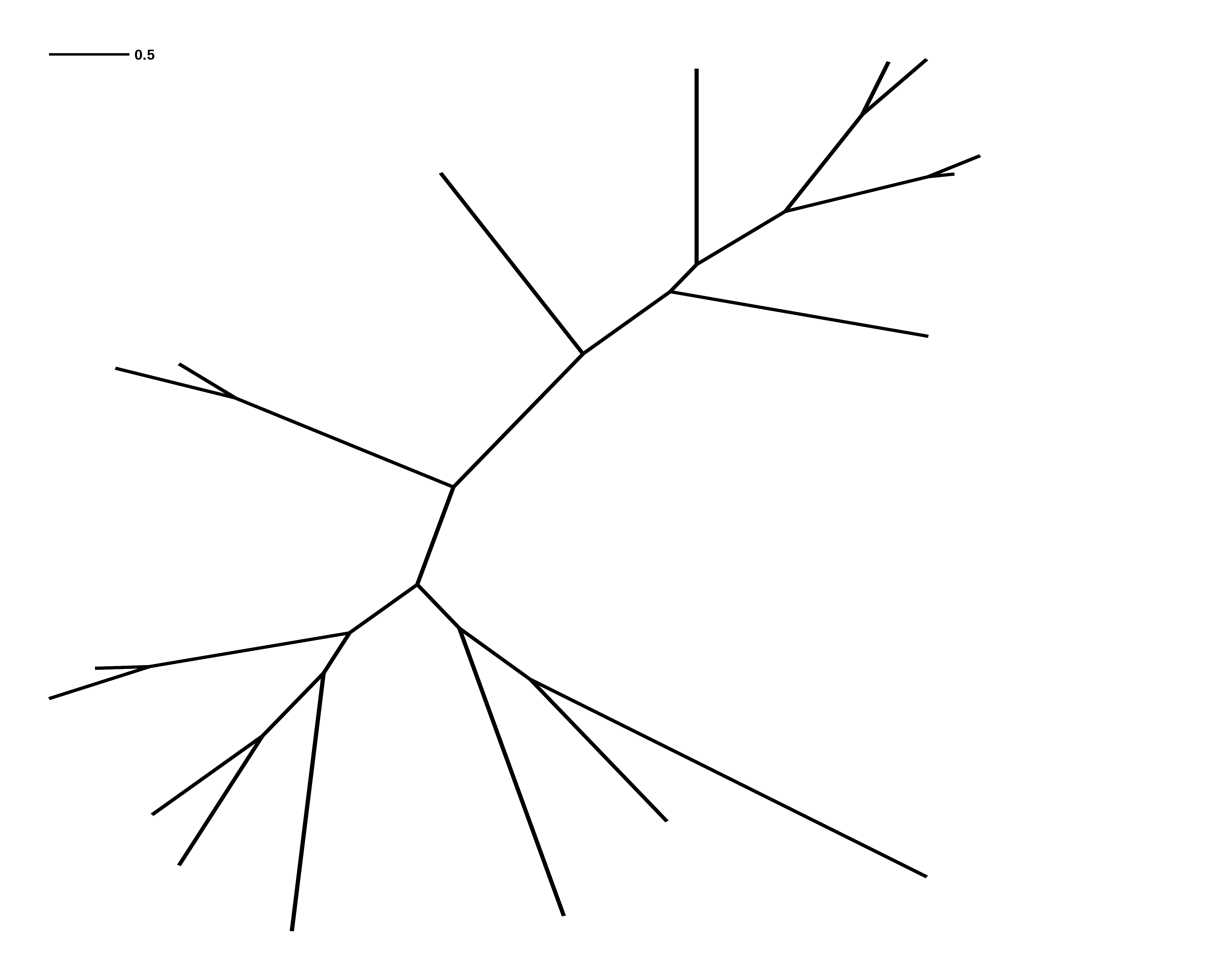
图1 无根树
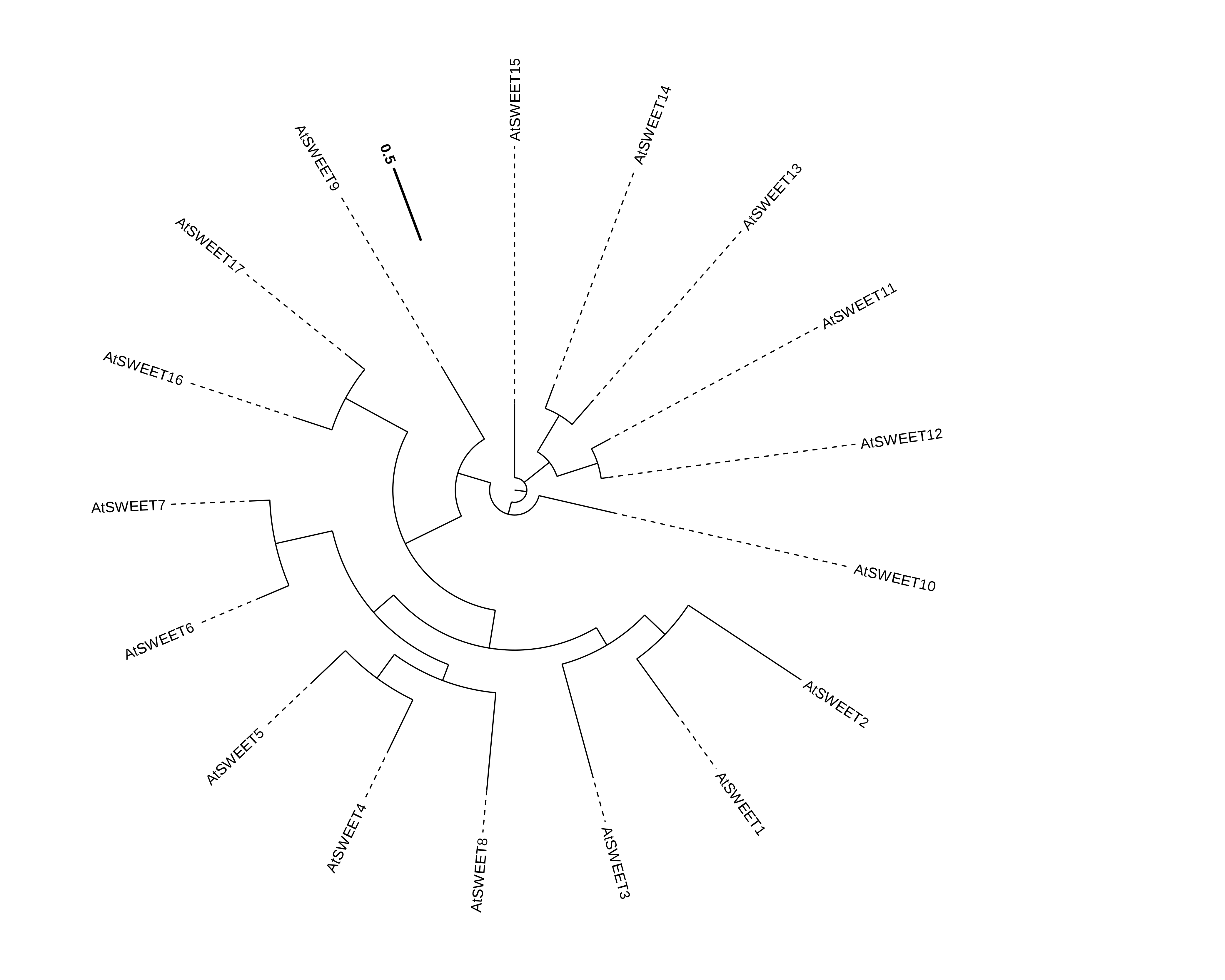
图2 圆形进化树
如果觉得该图形单调,可以使用iTOL(https://itol.embl.de/),美化结果如下图:
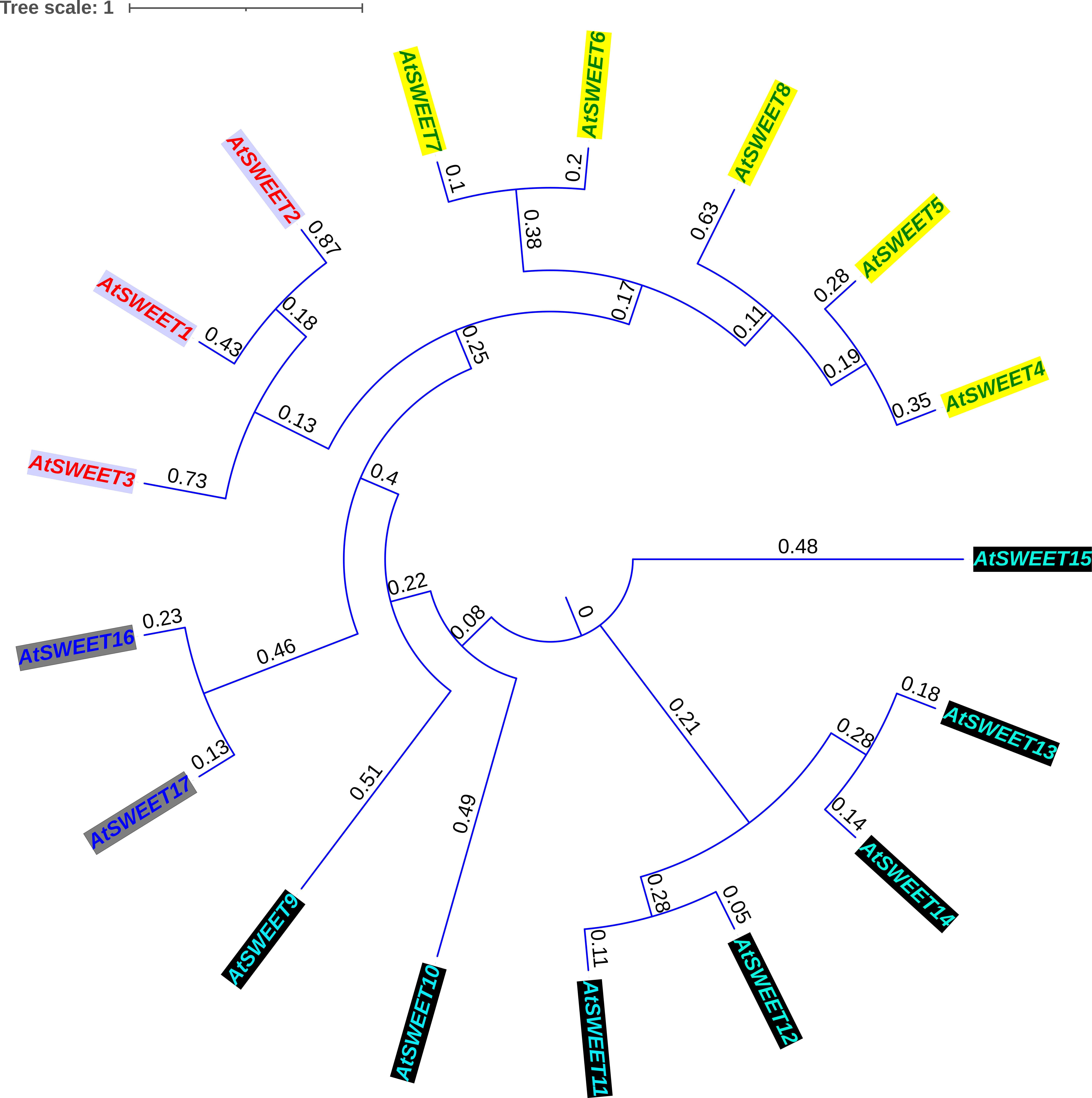
图2 输入.nwk文件,在iTOL中美化的圈图进化树
因为SWEET家族有4个亚家族,图中用4种不同的颜色来区分4个亚家族。
Q1. 上传的数据需要保存成什么格式?文件名称和拓展名有没有要求?
OmicShare当前支持txt(制表符分隔)文本文件、csv(逗号分隔)文本文件、以及Excel专用的xlsx格式,同样支持旧版Excel的xls(Excel 97-2003 )格式。如果是核酸、蛋白序列文件,必须为FASTA格式(本质是文本文件)。
文件名可由英文和数字构成,文件拓展名没有限制,可以是“.txt”、“.xlsx”、“.xls”、“.csv”“.fasta”等,例如 mydata01.txt,gene02.xlsx 。
Q2. 提交时报错常见问题:
1.提交时显示X行X列空行/无数据,请先自查表格中是否存在空格或空行,需要删掉。
2.提交时显示列数只有1列,但表格数据不止1列:列间需要用分隔符隔开,先行检查文件是否用了分隔符。
其它提示报错,请先自行根据提示修改;如果仍然无法提交,可通过左侧导航栏的“联系客服”选项咨询OmicShare客服。
Q3. 提交的任务完成后却不出图该怎么办?
主要原因是上传的数据文件存在特殊符号所致。可参考以下建议逐一排查出错原因:
(1)数据中含中文字符,把中文改成英文;
(2) 数据中含特殊符号,例如 %、NA、+、-、()、空格、科学计数、罗马字母等,去掉特殊符号,将空值用数字“0”替换;
(3)检查数据中是否有空列、空行、重复的行、重复的列,特别是行名(一般为gene id)、列名(一般为样本名)出现重复值,如果有删掉。
排查完之后,重新上传数据、提交任务。如果仍然不出图,可通过左侧导航栏的“联系客服”选项咨询OmicShare客服。
Q4.下载的结果文件用什么软件打开?
OmicShare云平台的结果文件(例如,下图为KEGG富集分析的结果文件)包括两种类型:图片文件和文本文件。

图片文件:
为了便于用户对图片进行后期编辑,OmicShare同时提供位图(png)和矢量图(pdf、svg)两种类型的图片。对于矢量图,最常见的是pdf和svg格式,常用Ai(Adobe illustrator)等进行编辑。其中,svg格式的图片可用网页浏览器打开,也可直接在word、ppt中使用。
文本文件:
文本文件的拓展名主要有4种类型:“.os”、“.xls”、“.log”和“.txt”。这些文件本质上都是制表符分隔的文本文件,使用记事本、Notepad++、EditPlus、Excel等文本编辑器直接打开即可。结果文件中,拓展名为“.os”文件为上传的原始数据;“.xls”文件一般为分析生成的数据表格;“.log”文件为任务运行日志文件,便于检查任务出错原因。
Q5. 提交的任务一直在排队怎么办?
提交任务后都需要排队,1分钟后,点击“任务状态刷新”按钮即可。除了可能需运行数天的注释工具,一般工具数十秒即可出结果,如果超出30分钟仍无结果,请联系OS客服,发送任务编号给OmicShare客服,会有专人为你处理任务问题。
Q6. 结果页面窗口有问题,图表加载不出来怎么办?
尝试用谷歌浏览器登录OmicShare查看结果文件,部分浏览器可能不兼容。